No podrás conectarte si excedes diez intentos fallidos.
La Agencia SINC ofrece servicios diferentes dependiendo de tu perfil.
Selecciona el tuyo:
La Comisión Europea ha designado un medicamento huérfano para el tratamiento de la Anemia de Fanconi. Se trata de un vector lentiviral que contiene el gen de esta enfermedad. En la designación de este medicamento huérfano, obtenida por el Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) y el Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT), han colaborado Genoma España y la Fundación Botín, entre otras instituciones.
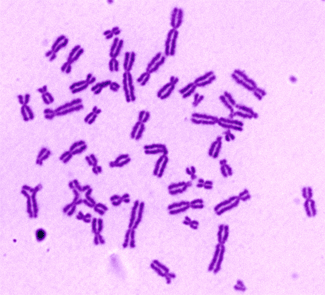
La Comisión Europea ha declarado en diciembre de 2010 el “Vector lentiviral conteniendo el gen de la anemia de Fanconi A (FANCA)” como un nuevo medicamento huérfano para el tratamiento de pacientes con anemia de Fanconi del tipo A.
El medicamento huérfano tiene por objeto corregir el defecto genético en las células madre hematopoyéticas de pacientes con anemia de Fanconi, de manera análoga a como se ha realizado en pacientes con otras enfermedades monogénicas, tales como algunas inmunodeficiencias primarias. Como novedad, el vector desarrollado pertenece a la familia de los lentivirus, de mayor seguridad y eficacia respecto a los vectores usados en los primeros protocolos de terapia génica.
Una vez corregido el defecto genético de las células madre hematopoyéticas, éstas serán reinfundidas en los pacientes, con objeto de rescatar y/o prevenir el fallo de médula ósea, que aparece en la inmensa mayoría de estos pacientes.
Los trabajos realizados para la designación de este medicamento huérfano han sido coordinados por el doctor Juan Bueren, Jefe de la División de Hematopoyesis y Terapia Génica del Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT) y Jefe de Grupo del Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), dependiente del Instituto de Salud Carlos III.
Colaboración de varias instituciones
En este Proyecto han colaborado principalmente Genoma España (Proyecto FANCOGENE), la Comisión Europea (6º y 7º Programa Marco), el Ministerio de Ciencia e Innovación (Programas Nacionales de Investigación) y la Fundación Botín, así como otras Instituciones que han colaborado en la financiación del Proyecto FANCOGENE, entre las que se incluye la Asociación Española de Anemia de Fanconi, el Centro Nacional de Investigaciones Oncológicas, la Universidad Autónoma de Barcelona y las empresas Genzyme y Pharmamar S.A.
La anemia de Fanconi es una enfermedad hereditaria de baja prevalencia (unos 150 pacientes en España), que afecta principalmente a la médula ósea, disminuyendo todos los tipos de células sanguíneas. La falta de glóbulos blancos predispone al paciente a infecciones, mientras que la falta de plaquetas y glóbulos rojos puede producir sangrado y fatiga, respectivamente.
El tratamiento actual se basa principalmente en el trasplante de células de la médula ósea de un donante compatible, preferentemente un hermano del paciente. El medicamento huérfano desarrollado por el CIBERER/CIEMAT está fundamentalmente dirigido a aquellos pacientes que no tienen un donante adecuado, y por tanto con escasas alternativas terapéuticas.
Un nuevo estudio revela que las personas que soportan abusos y desatención en la infancia pueden tener tres veces más probabilidades de ingresar en un hospital por trastornos relacionados con el consumo de alcohol y otras sustancias a la edad de 40 años, en comparación con las que no sufren estos daños.
Un estudio, llevado a cabo por investigadores del Instituto de Salud Carlos III, concluye que la introducción en nuestro país en 2015 de la inmunización de embarazadas frente a esta enfermedad ha permitido reducir las tasas de hospitalización y la duración de los ingresos hospitalarios en menores de 3 meses.



